



- 商品介绍
- 规格参数
- 包装参数
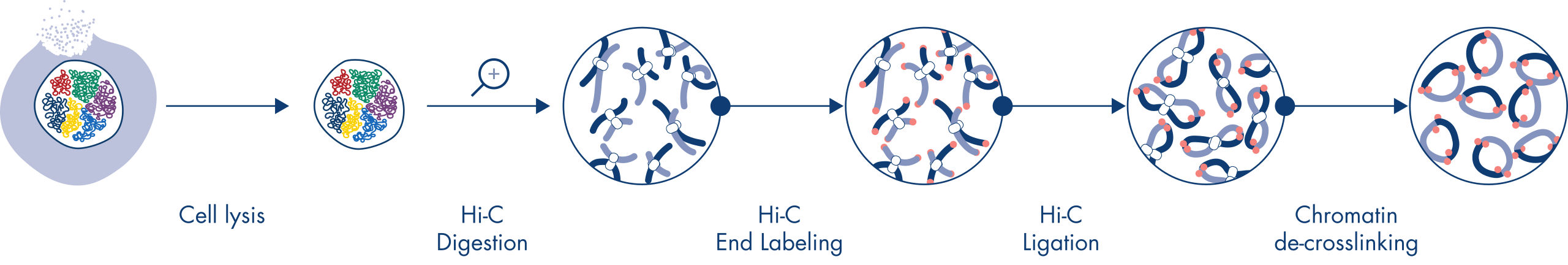
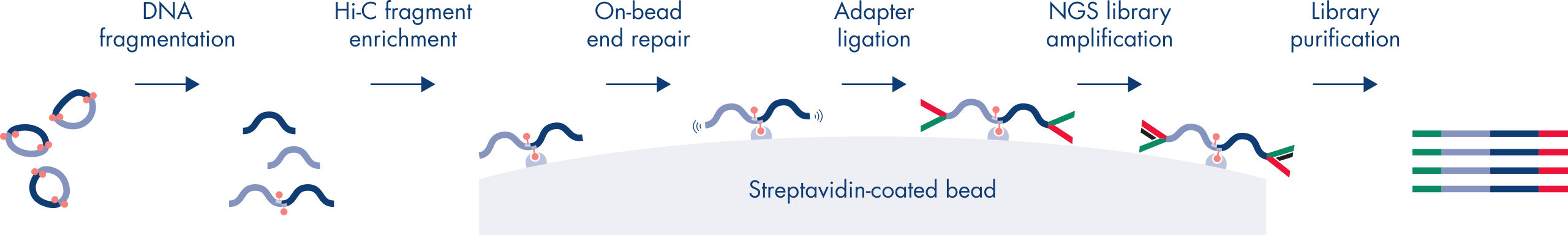
- All-inclusive kit – quality-controlled reagents for generating Hi-C NGS libraries
- Included Illumina adapters with sequence bar codes for multiplex sequencing
- Fully tested, robust and fast protocol – Hi-C library generation in <2 days
- Low sample input requirement – libraries generated from just 5000 cells
- Data analysis pipeline based on open-source tools
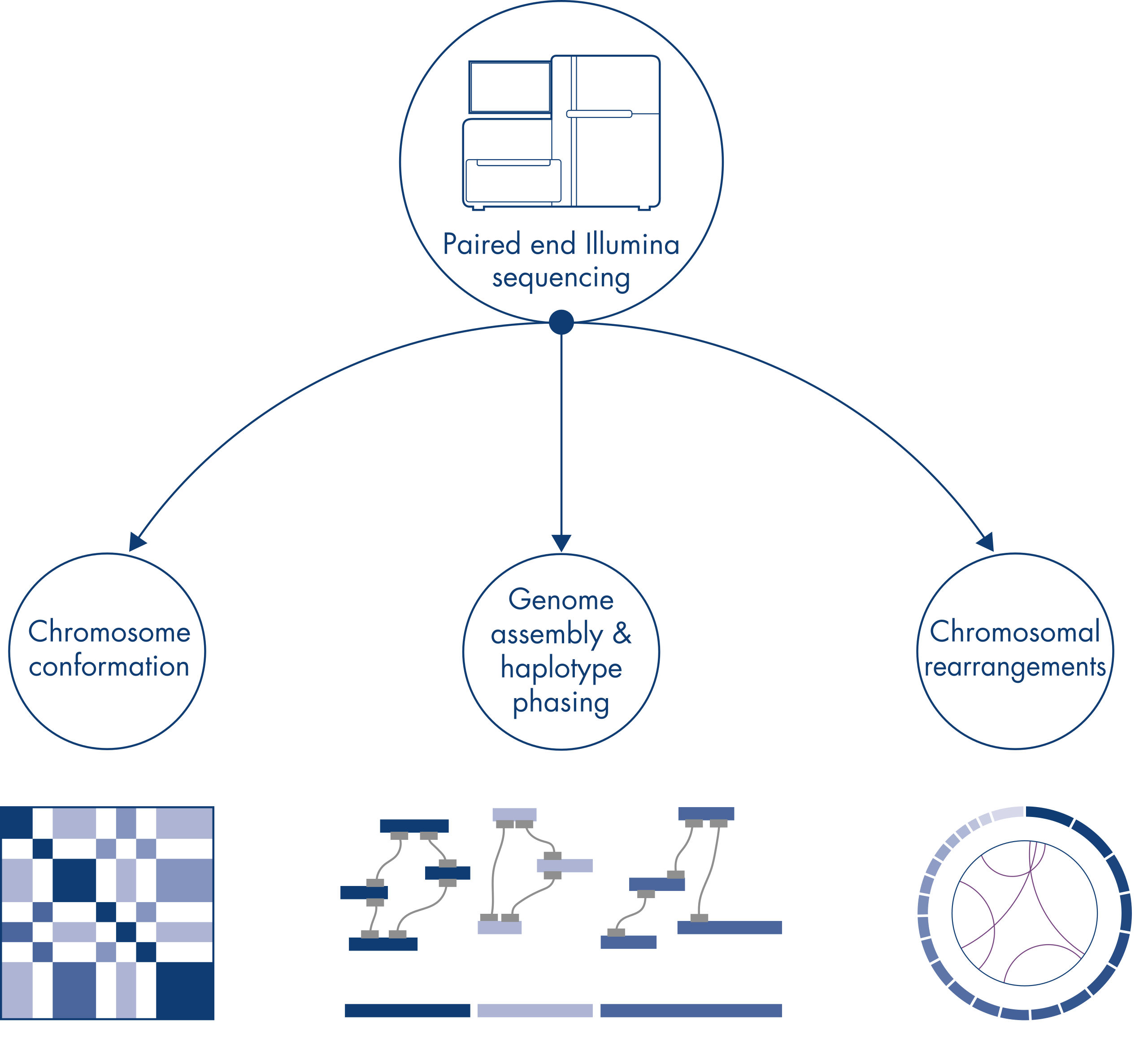
Chromatin conformation
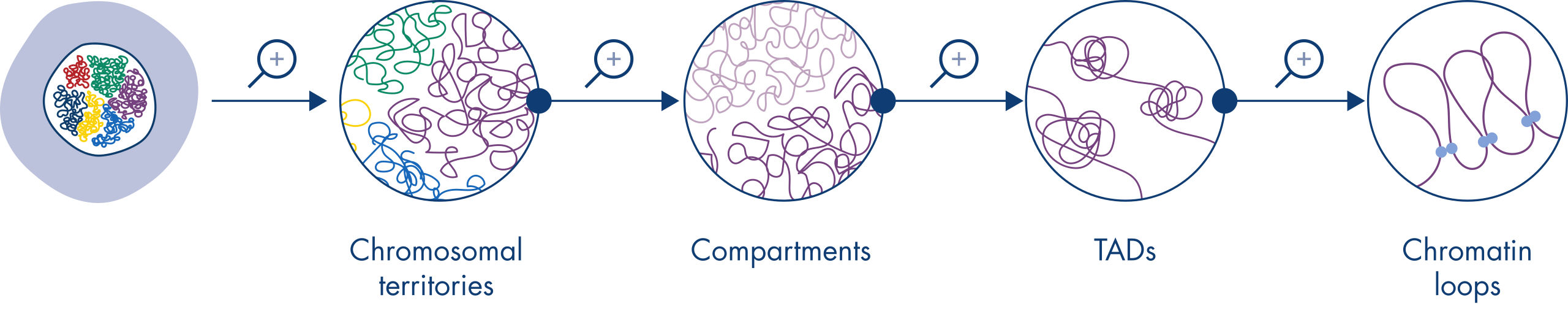
Hi-C has quickly become a very important tool for the analysis of nuclear organization. Analysis of Hi-C data has revealed the amazing complexity of genome architecture, with multiple layers of spatial organization that partition the genome into chromosome territories, chromosomal sub-compartments, topologically associated domains (TADs) and DNA loops at increasing resolution (see figure Levels of chromatin organization
Chromosomal rearrangements and copy number variants
Individual chromosomes are physically separated into discrete territories and, therefore, DNA interactions captured by Hi-C primarily take place between DNA from the same chromosome (in cis) with little interaction between chromosomes (in trans). Owing to this phenomenon, Hi-C can be used as a genome-wide assay to identify translocations and other structural variants of interest. Compared to other NGS techniques, Hi-C requires extremely low coverage, which can save on costs. Furthermore, rearrangements involving poorly mappable regions can be better detected using Hi-C than standard NGS methods. Conveniently, the same Hi-C data can be used to detect copy number changes.
Genome assembly – haplotype phasing
When sequencing and assembling the genomes of new species, generating sequence scaffolds is often limited by large stretches of repetitive sequences that extend beyond the range of sequencing. In Hi-C data, the vast majority of interactions occur in cis between loci on the same chromosome. Additionally, a significant portion of these cis interactions is long-range, occurring between loci separated by millions of bases of DNA. These properties of chromatin interactions can be leveraged to order, orient and join sequence scaffolds into near full-length chromosomes without the need for a reference genome. Using the same principles, Hi-C interaction maps can be used to create diploid genomes by assigning genetic variants to paternal and maternal sister chromosomes (see figure Downstream applications of Hi-C sequencing data
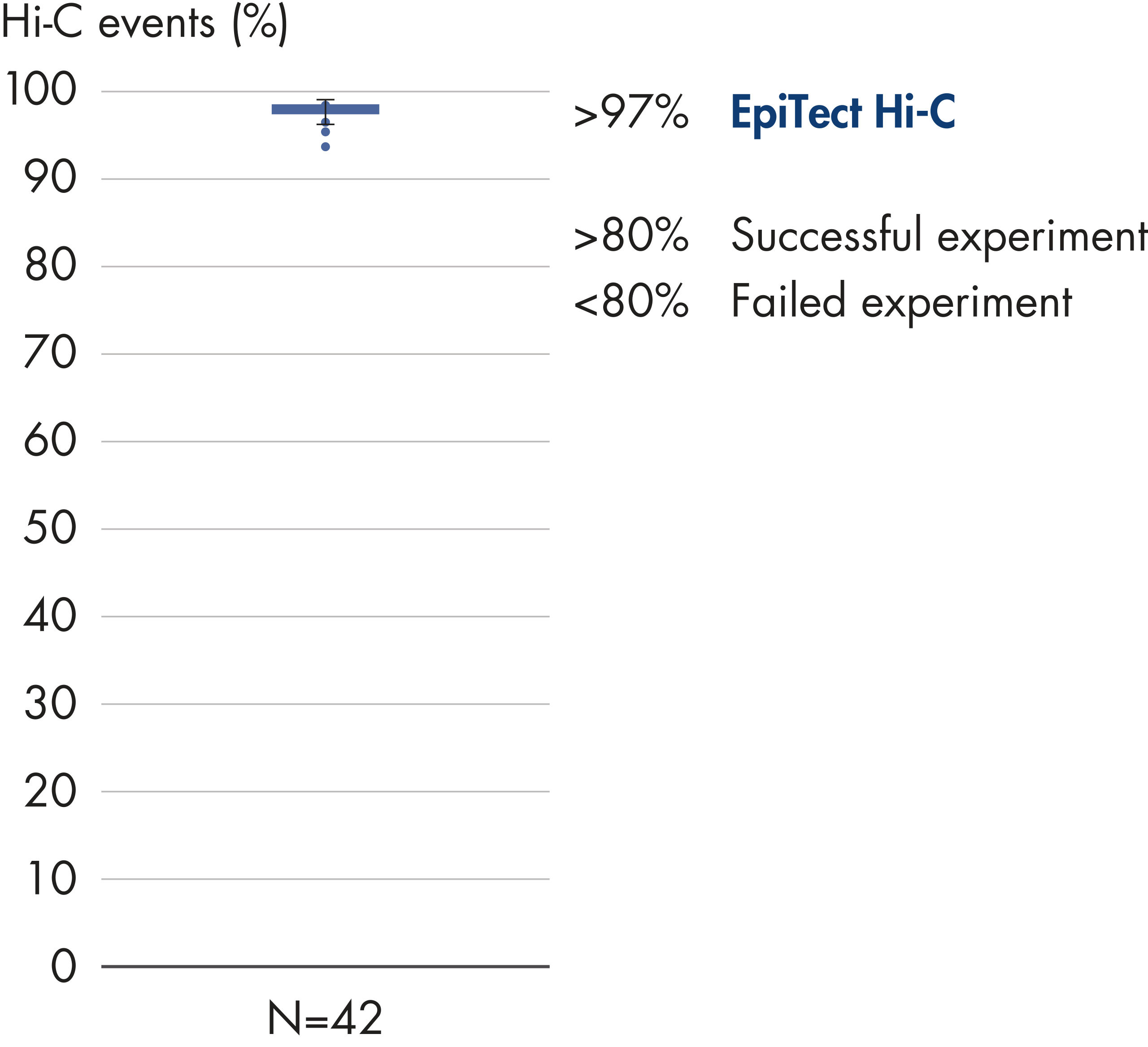
Percentage of Hi-C events
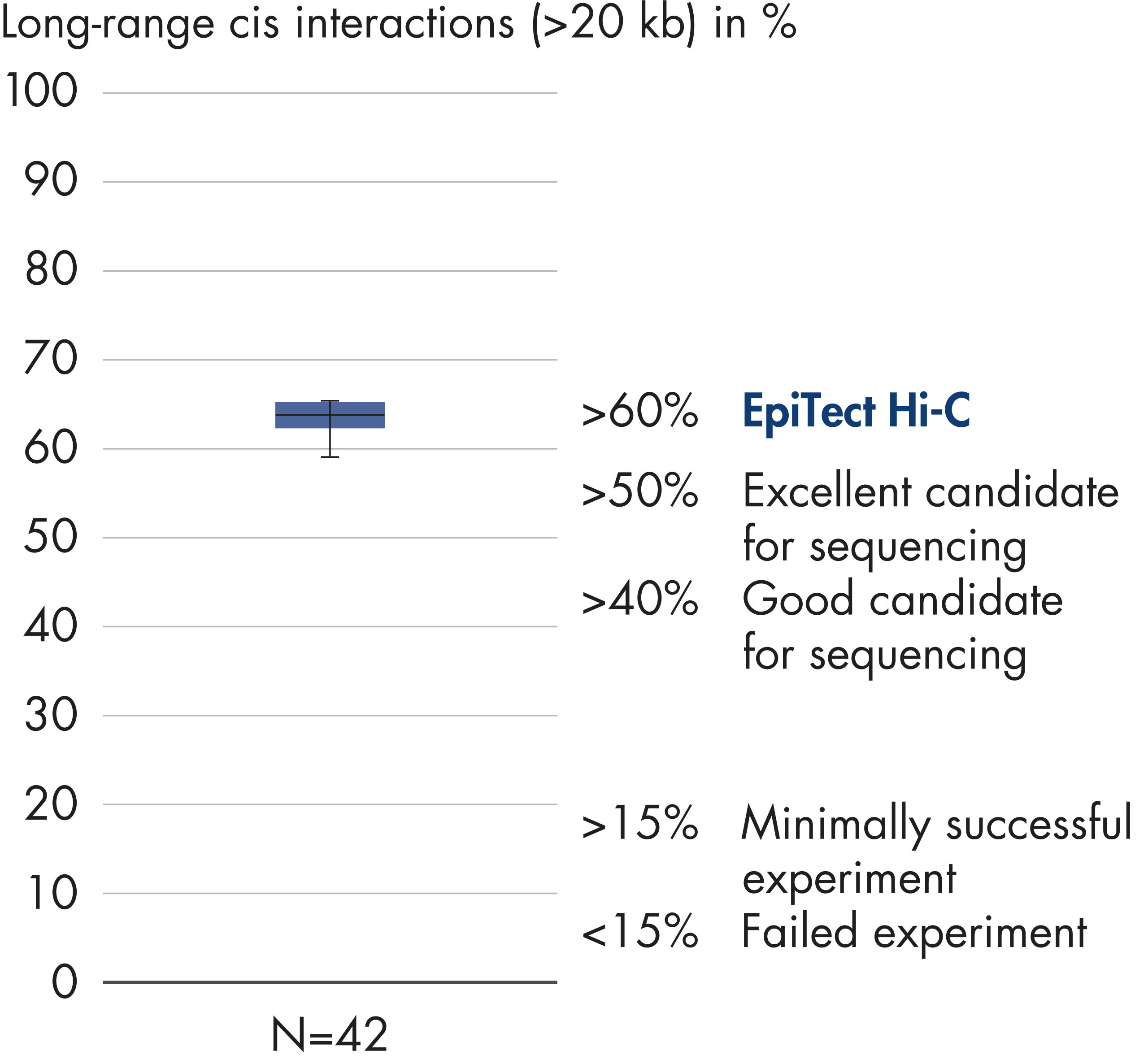
Percentage of long-range cis interactions
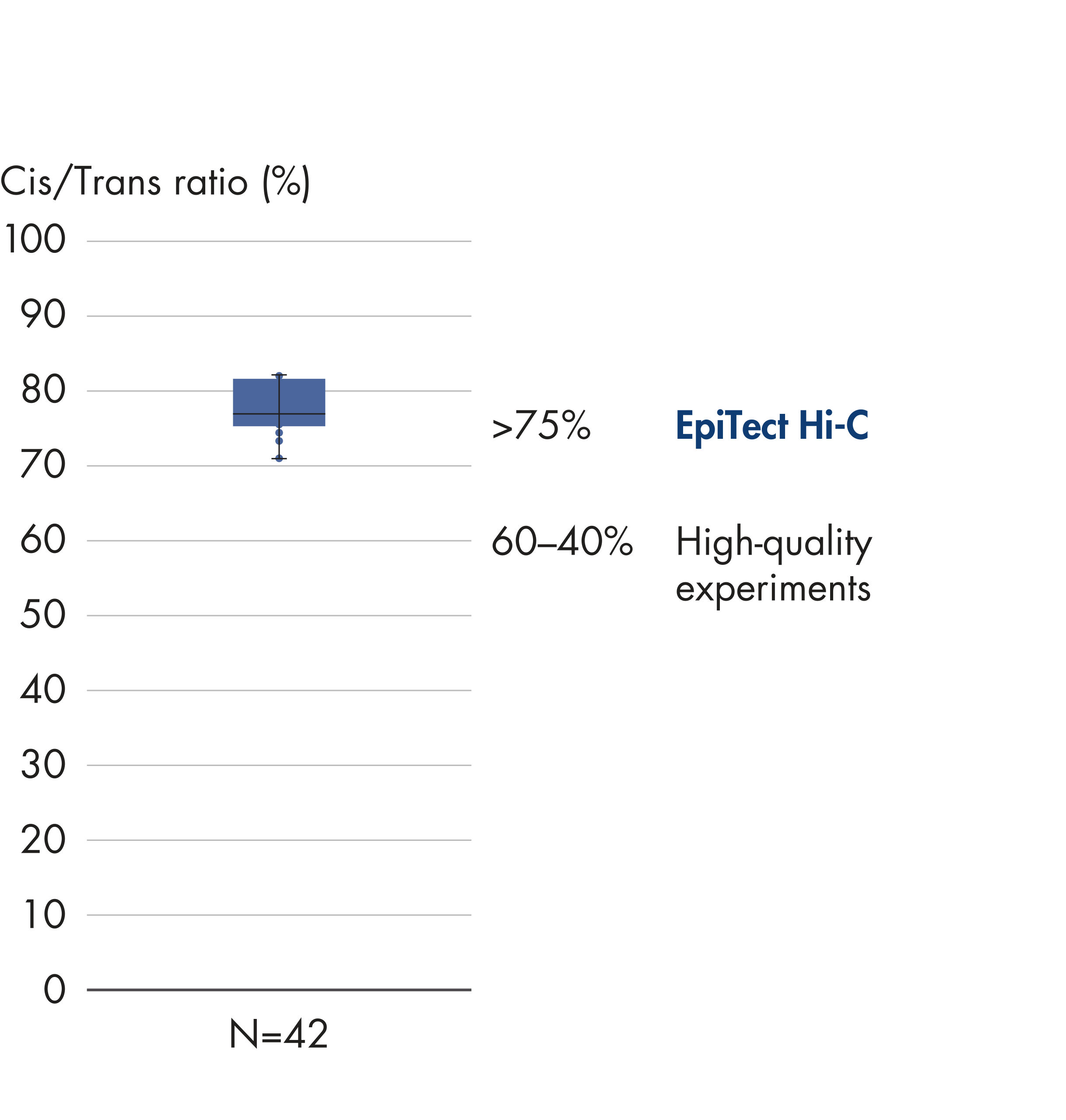
Cis/Trans ratio
No strand orientation bias with the EpiTect Hi-C Kit
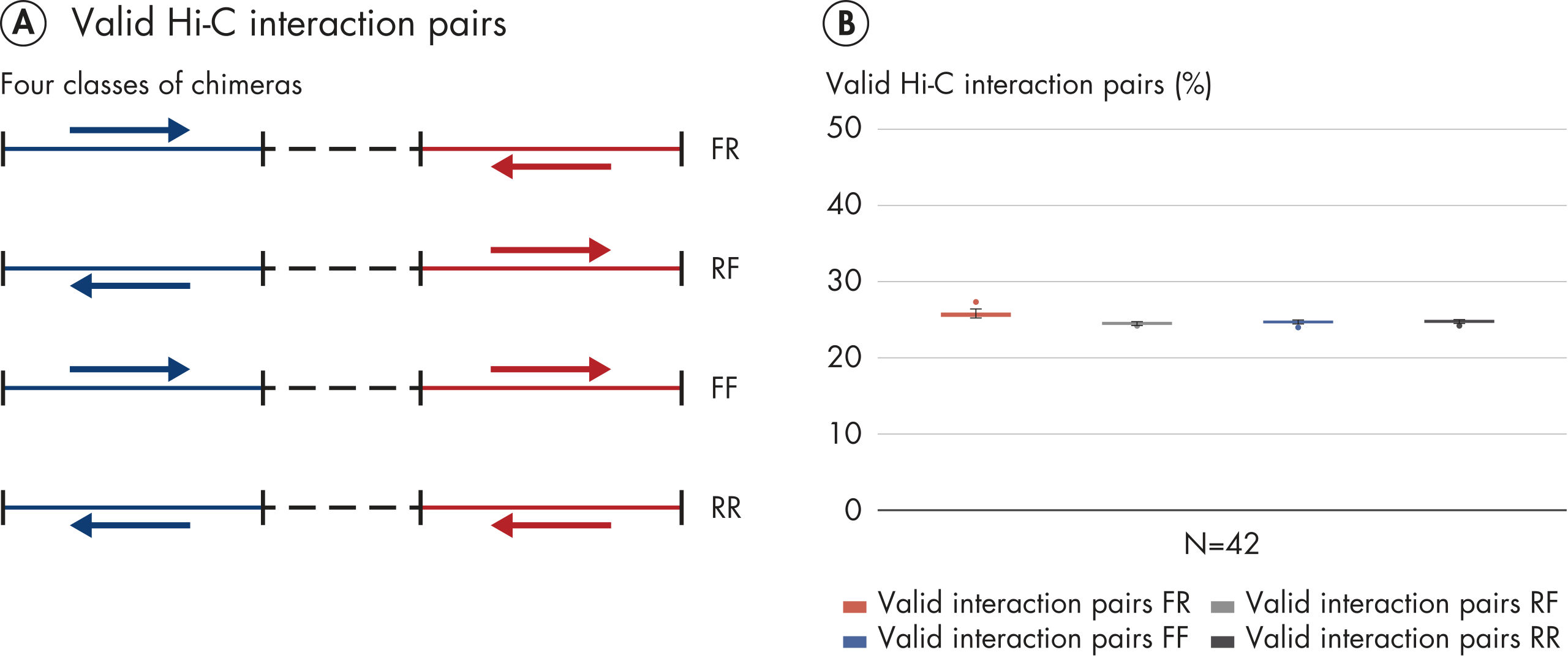
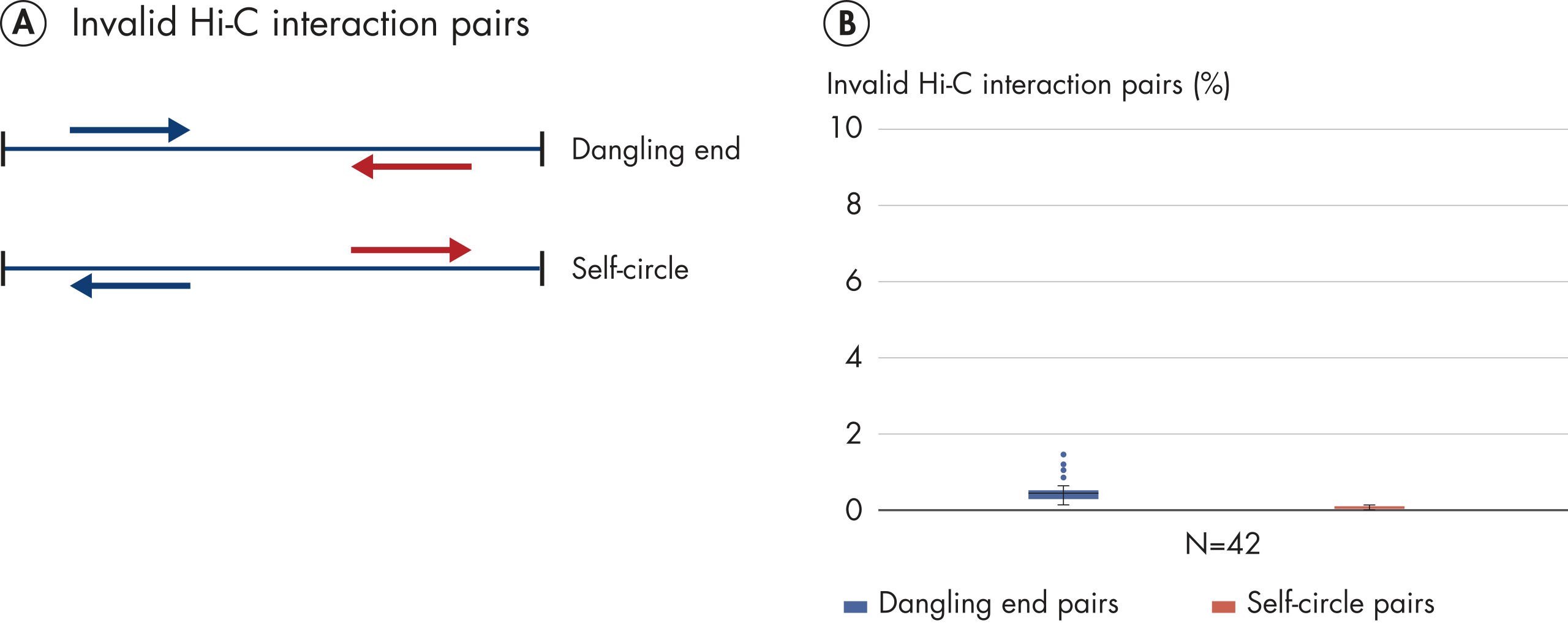
Hi-C proximity ligation of chromatin generates interaction pairs in the form of DNA chimeras. When valid Hi-C interactions pairs (DNA chimeras) are paired-end sequenced, read 1 (blue arrow) and read 2 (red arrow) will map to 2 different restriction fragments (A: blue line and red line). Hi-C chimeras can be divided into 4 classes, distinguished by the strand orientation of read pairs as shown in A. If the chimeras are a result of random proximity ligation of chromatin, then 25% of each class of chimera is expected. The absence of strand bias observed with EpiTect Hi-C NGS libraries, as shown in B, is further evidence of the high performance of the EpiTect Hi-C Kit.
;白色高密度聚乙烯螺旋盖,500ml容量")





















 微信小程序
7X24小时在线咨询
微信小程序
7X24小时在线咨询
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}